
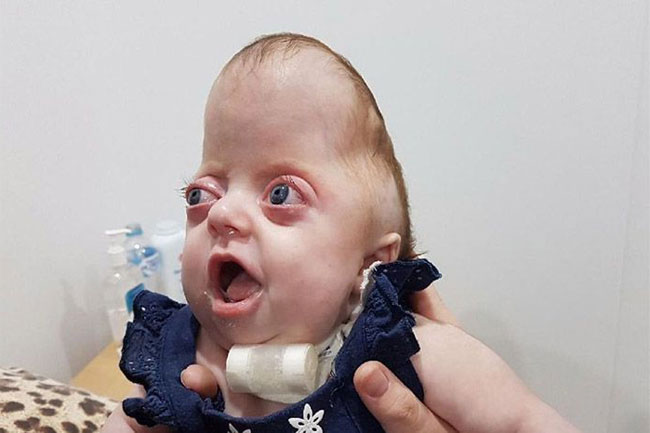
Das Pfeiffer-Syndrom tritt auf, wenn die Knochen im Schädel, in den Händen und Füßen Ihres Kindes aufgrund einer Genmutation zu früh im Mutterleib verschmolzen sind. Dies kann zu körperlichen, geistigen und inneren Symptomen führen.
Das Pfeiffer-Syndrom ist extrem selten. Nur etwa eines von 100.000 Kindern wird damit geboren.
Das Pfeiffer-Syndrom wird oft erfolgreich behandelt. Lesen Sie weiter, um mehr über das Pfeiffer-Syndrom zu erfahren, was es verursacht und wie Ihr Kind behandelt werden kann.
Welche Arten von dieser Erkrankung gibt es?
Es gibt drei Arten des Pfeiffer-Syndroms.
Typ 1
Typ 1 ist die mildeste und bei weitem die häufigste Form dieses Syndroms. Ihr Kind wird einige körperliche Symptome haben, aber normalerweise keine Probleme mit seiner Gehirnfunktion. Kinder mit diesem Typ können leben, um ein Erwachsener mit wenigen Komplikationen zu sein.
Ihr Kind kann mit einigen der folgenden Symptome geboren werden:
- okulärer Hypertelorismus oder Augen, die weit auseinander liegen.
- Stirn, die hoch auf dem Kopf aussieht und sich durch die früh verschmelzenden Schädelknochen ausdehnt.
- Brachyzephalie oder Flachheit des Hinterkopfes
- Unterkiefer, der herausragt
- hypoplastischer Oberkiefer oder ein Oberkiefer, der sich noch nicht vollständig entwickelt hat.
- breite, große Daumen und große Zehen, die sich von den anderen Fingern und Zehen weg erstrecken.
- Hörschwäche
- Zahn- oder Zahnfleischprobleme haben
Typ 2
Kinder mit Typ 2 können eine oder mehrere schwere oder lebensbedrohliche Formen von Typ-1-Symptomen aufweisen. Möglicherweise muss Ihr Kind operiert werden, damit es bis ins Erwachsenenalter überlebt.
Andere Symptome sind:
- Kopf- und Gesichtsknochen sind früh verschmolzen und bilden eine Form, die als „Kleeblatt“ bezeichnet wird.
- Proptose oder Exophthalmus, was passiert, wenn die Augen Ihres Kindes aus den Augenhöhlen ragen.
- Verzögerungen in der Entwicklung oder Lernschwierigkeiten, da die frühe Fusion der Schädelknochen das Gehirn Ihres Kindes daran gehindert hat, vollständig zu wachsen.
- Fusion in anderen Knochen, wie z.B. den Ellenbogen- und Kniegelenken, genannt Ankylose.
- Unfähigkeit, aufgrund von Problemen mit der Luftröhre, dem Mund oder der Nase richtig zu atmen.
- Probleme mit dem Nervensystem, wie z.B. Flüssigkeit aus der Wirbelsäule, die sich im Gehirn sammelt, genannt Hydrozephalie.
Typ 3
Ihr Kind kann schwere oder lebensbedrohliche Formen der Symptome der Typen 1 und 2 haben. Sie werden keinen Kleeblattschädel haben, können aber Probleme mit ihren Organen wie Lunge und Nieren haben.
Eine frühzeitige Schädelknochenfusion kann zu Lern- oder kognitiven Behinderungen führen. Ihr Kind muss möglicherweise ein Leben lang umfassend operiert werden, um diese Symptome zu behandeln und bis ins Erwachsenenalter zu überleben.
Was verursacht diesen Zustand?
Das Pfeiffer-Syndrom entsteht, weil die Knochen, die den Schädel, die Hände oder die Füße Ihres Kindes bilden, zu früh miteinander verschmelzen, während sie sich in der Gebärmutter befinden. Dies ist es, was dazu führt, dass Ihr Kind mit einem ungewöhnlich geformten Schädel oder mit Fingern und Zehen geboren wird, die weiter auseinander gespreizt sind als sonst.
Dies kann wenig Platz für das Gehirn oder andere Organe lassen, um bis zum Anschlag zu wachsen, was zu Komplikationen führen kann:
- kognitive Funktion
- Atmung
- andere wichtige Körperfunktionen, wie Verdauung oder Bewegung
Wer bekommt diese Bedingung?
Das Pfeiffer-Syndrom vom Typ 1 wird durch eine Mutation in einem oder mehreren Genen verursacht, die Teil der Knochenentwicklung eines Kindes sind.
Nur ein Elternteil muss das Gen tragen, um es an sein Kind weiterzugeben, so dass sein Kind eine 50-prozentige Chance hat, die Krankheit zu erben. Dies wird als autosomal dominantes Muster bezeichnet. Das Pfeiffer-Syndrom vom Typ 1 kann entweder auf diese Weise vererbt werden oder durch eine neue Genmutation entstehen.
Die Forschung deutet darauf hin, dass Typ 1 durch eine Mutation in einem der beiden Fibroblasten-Wachstumsfaktor-Rezeptorgene, FGFR1 oder FGFR2, verursacht wird. Die Typen 2 und 3 werden fast immer durch eine Mutation im FGFR2-Gen verursacht und sind neue (spontane) Mutationen und nicht vererbt.
Die Forschung deutet auch darauf hin, dass Spermien von älteren Männern eher mutieren. Dies kann dazu führen, dass das Pfeiffer-Syndrom, insbesondere die Typen 2 und 3, ohne Vorwarnung auftritt.
Wie wird dieser Zustand diagnostiziert?
Ihr Arzt kann manchmal das Pfeiffer-Syndrom diagnostizieren, während Ihr Kind noch in der Gebärmutter ist, indem er mit Hilfe von Ultraschallbildern eine frühe Fusion der Schädelknochen und Symptome der Finger und Zehen Ihres Kindes erkennt.
Wenn es sichtbare Symptome gibt, wird Ihr Arzt in der Regel bei der Geburt Ihres Kindes eine Diagnose stellen. Wenn die Symptome Ihres Kindes leicht sind, kann es sein, dass Ihr Arzt die Krankheit erst einige Monate oder sogar Jahre nach der Geburt Ihres Kindes und dem Beginn des Wachstums diagnostiziert.
Ihr Arzt kann Ihnen, Ihrem Partner und Ihrem Kind vorschlagen, Gentests durchzuführen, um nach Mutationen an den FGFR-Genen zu suchen, die das Pfeiffer-Syndrom verursachen, und um zu sehen, wer das Gen trägt.
Wie wird diese Erkrankung behandelt?
Etwa drei Monate nach der Geburt Ihres Kindes empfiehlt Ihr Arzt in der Regel eine Operation in mehreren Phasen, um den Schädel Ihres Kindes umzugestalten und den Druck auf das Gehirn zu verringern.
Zunächst werden die Lücken zwischen den verschiedenen Knochen im Schädel Ihres Kindes, die so genannten synostotischen Nähte, getrennt. Dann wird der Schädel rekonstruiert, so dass das Gehirn Platz zum Wachsen hat und der Schädel eine symmetrischere Form annehmen kann. Ihr Arzt wird Ihnen auch einen langfristigen Behandlungsplan geben, um sicherzustellen, dass die Zähne Ihres Kindes gepflegt werden.
Sobald Ihr Kind von diesen Operationen geheilt ist, kann Ihr Arzt auch eine Langzeitoperation vorschlagen, um die Symptome von Kiefer, Gesicht, Händen oder Füßen so zu behandeln, dass sie atmen können, sowie ihre Hände und Füße zum Bewegen verwenden.
Möglicherweise muss Ihr Arzt kurz nach der Geburt Ihres Kindes eine Notoperation durchführen, damit Ihr Kind durch Nase oder Mund atmen kann. Sie sorgen auch dafür, dass Herz, Lunge, Magen und Nieren normal funktionieren.
Leben mit dem Pfeiffer-Syndrom
Es besteht eine gute Chance, dass Ihr Kind mit anderen Kindern spielen, zur Schule gehen und mit dem Pfeiffer-Syndrom bis ins Erwachsenenalter überleben kann. Das Pfeiffer-Syndrom vom Typ 1 ist durch frühzeitige Operation, Physiotherapie und langfristige Operationsplanung behandelbar.
Typ 2 und 3 kommen nicht sehr oft vor. Wie Typ 1 können sie oft mit einer Langzeitoperation und der Rekonstruktion des Schädels, der Hände, Füße und anderer Knochen und Organe Ihres Kindes behandelt werden, die betroffen sein können.
Die Aussichten für Kinder mit Typ 2 und 3 sind nicht so gut wie die für Typ 1. Dies liegt an den Auswirkungen, die die frühe Fusion der Knochen Ihres Kindes auf das Gehirn, die Atmung und die Bewegungsfähigkeit haben kann.
Eine frühzeitige Behandlung, zusammen mit einer lebenslangen körperlichen und geistigen Rehabilitation und Therapie, kann Ihrem Kind helfen, als Erwachsener zu leben, mit nur einigen Komplikationen, die seine kognitive Funktion und Mobilität betreffen.
